
KIMOZO 40 mg-ml, suspension buvable, boîte de 1 flacon ( seringue doseuse graduée en ml) de 20 ml
Retiré du marché le : 08/08/2025
Dernière révision : 27/04/2022
Taux de TVA : 0%
Laboratoire exploitant : ORPHELIA PHARMA SAS
Source :

Kimozo est indiqué en monothérapie ou en association à un inhibiteur spécifique de l'ADN topoisomérase I (irinotécan ou topotecan) dans le traitement des patients pédiatriques âgés de 1 à 6 ans et chez les patients âgés de plus de 6 ans dans l'incapacité d'avaler le témozolomide sous forme de gélule et atteints :
- d'un neuroblastome à haut risque réfractaire ou présentant une réponse insuffisante à la chimiothérapie d'induction.
- d'un neuroblastome à haut risque récidivant après une réponse au moins partielle à la chimiothérapie d'induction suivie d'un traitement myéloablatif et d'une greffe de cellules souches.
-
se référer à la rubrique Propriétés pharmacodynamiques
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique Liste des excipients.
Hypersensibilité à la dacarbazine (DTIC).
Myélosuppression sévère (voir rubrique Mises en garde spéciales et précautions d'emploi).
L'expérience clinique de l'utilisation du témozolomide chez les enfants et les adolescents est globalement plus limitée que chez les adultes.
Bien que les données soient limitées, le profil de tolérance chez les enfants est attendu similaire à celui de l'adulte et les mises en garde spéciales et précautions d'emploi s'appuient sur celles de l'utilisation du témozolomide chez l'adulte.
Infections opportunistes et réactivation d'infections
Des infections opportunistes (telle que la pneumonie à Pneumocystis jirovecii) et la réactivation d'infections (telles que VHB et CMV) ont été observées pendant le traitement par le témozolomide (voir rubrique Effets indésirables).
Méningo-encéphalite herpétique
Une méningo-encéphalite herpétique (ayant parfois entraîné une issue fatale) a été observée chez des patients recevant du témozolomide en association avec une radiothérapie, notamment en cas d'administration concomitante de stéroïdes.
Pneumomie à Pneumocystis jirovecii
Dans un essai pilote, les patients ayant reçu du témozolomide de manière concomitante à la radiothérapie sur un schéma prolongé de 42 jours ont montré un risque particulier de développer des
pneumonies à Pneumocystis jirovecii (PPJ).Il peut y avoir une fréquence plus importante de PPJ quand le témozolomide est administré selon un schéma de traitement plus long. Néanmoins, tous les patients recevant du témozolomide, particulièrement les patients recevant aussi des stéroïdes, doivent être surveillés attentivement concernant le développement de PPJ, quel que soit le schéma de traitement. Des cas d'insuffisance respiratoire fatale ont été rapportés chez des patients utilisant du témozolomide, en particulier en association avec la dexaméthasone ou d'autres stéroïdes.
Il est recommandé d'initier une prophylaxie de la pneumonie à Pneumocystis jirovecii pour les patients recevant du témozolomide.
VHB
Des cas d'hépatite liée à une réactivation du virus de l'hépatite B (VHB) ont été rapportés, parfois d'évolution fatale. Les patients présentant une sérologie positive pour l'hépatite B (incluant les patients présentant une hépatite B active) doivent être adressés à un médecin spécialisé en hépatologie avant l'instauration du traitement. Au cours du traitement, les patients devront être surveillés et pris en charge de façon appropriée.
Hépatotoxicité
Des cas d'atteintes hépatiques, dont des cas d'insuffisance hépatique fatale, ont été rapportés chez des patients traités par du témozolomide (voir rubrique Effets indésirables). Avant l'initiation du traitement, des examens de la fonction hépatique devront être réalisés pour obtenir des valeurs de référence. En cas d'anomalies, les médecins devront évaluer le rapport bénéfice/risque incluant le risque d'insuffisance hépatique fatale, avant d'initier le traitement par le témozolomide. Les examens de la fonction hépatique devront être réalisés après chaque cycle de traitement chez tous les patients. Chez les patients ayant des perturbations significatives de la fonction hépatique, les médecins devront évaluer le rapport bénéfice/risque de la poursuite du traitement. Une toxicité hépatique peut survenir plusieurs semaines, voire plus, après la dernière prise de témozolomide.
Tumeurs malignes
Des cas de syndromes myélodysplasiques et de tumeurs malignes secondaires, incluant la leucémie myéloïde, ont également été observés, très rarement (voir rubrique Effets indésirables).
Traitement antiémétique
Les nausées et les vomissements sont très fréquemment associés au témozolomide.
Un traitement antiémétique peut être administré avant ou après administration de Kimozo.
Paramètres biologiques
Chez les patients traités avec témozolomide, une myélosuppression peut survenir, y compris une pancytopénie prolongée pouvant entraîner une anémie aplasique qui dans certains cas, a eu une issue fatale. Dans certains cas, l'exposition concomitante à des médicaments pouvant être à l'origine d'une anémie aplasique, y compris la carbamazépine, phénytoïne et sulfamethoxazole/triméthoprime, complique l'évaluation.
Avant toute administration, les paramètres biologiques doivent être les suivants : taux de PNN ≥ 0,75 x 109/l et taux de plaquettes ≥ 75 x 109/l. S'il y a une atteinte avérée de la moelle osseuse, les paramètres biologiques suivants peuvent être considérés comme acceptables : PNN ≥ 0,50 x 109/l et plaquettes ≥ 50 x 109/l. Pendant le traitement, plusieurs bilans biologiques pourront être effectués, dont une numération formule sanguine à réaliser dans les 3 jours précédant l'initiation du cycle suivant. L'initiation d'un nouveau cycle de traitement sera retardée tant que les taux suivants ne seront pas atteints : taux de PNN > 0.75 x 109/l et taux de plaquettes > 75 x 109/l (si atteinte avérée de la moelle osseuse, PNN ≥ 0,50 x 109/l et plaquettes ≥ 50 x 109/l). Les retards d'initiation de cycle, les diminutions de dose et les arrêts selon l'utilisation du témozolomide en monothérapie ou en association avec topotécan ou irinotécan doivent être effectués selon les tableaux fournis en rubrique Posologie et mode d'administration.
Lésions buccales
Les patients sous chimiothérapie sont susceptibles de développer des lésions buccales de type stomatite/mucite. L'utilisation du témozolomide peut entrainer des lésions buccales (voir rubrique Effets indésirables). Compte tenu de la forme pharmaceutique spécifique de Kimozo (suspension buvable) et par précaution d'utilisation, il est recommandé de rincer la bouche avec une gorgée d'eau après l'administration du produit pour éviter tout contact prolongé de la muqueuse buccale avec des quantités résiduelles de témozolomide.
Teneur en sodium
Benzoate de sodium
Ce médicament contient 1 mg/ml de benzoate de sodium (E211).
Le profil de sécurité de Kimozo est basé sur les données rapportées depuis la mise sur le marché du témozolomide sous différentes présentations pharmaceutiques (solution pour perfusion, gélules) pour une utilisation dans des populations essentiellement adultes. Bien que les données soient limitées, la tolérance du témozolomide chez les enfants est attendue similaire à celle de l'adulte.
Résumé du profil de sécurité
Expérience issue d'essais cliniques
Chez les patients adultes traités avec du témozolomide dans les essais cliniques, les effets indésirables les plus fréquents étaient : nausées, vomissements, constipation, anorexie, céphalées, fatigue, convulsions et rash. La plupart des effets indésirables hématologiques a été rapportée fréquemment.
Le témozolomide présente globalement le même profil de sécurité dans le traitement du neuroblastome, que dans le traitement des tumeurs cérébrales pour lequel il a été originellement développé. Les effets indésirables hématologiques de Grade 3 et 4 (anémie et surtout neutropénie, thrombocytopénie) sont fréquemment rapportés (jusqu'à 10% des patients) et constituent la principale cause de retard de cycle, d'adaptation de dose ou d'arrêt de traitement. Le traitement combinant témozolomide avec les inhibiteurs de topoisomérase I semble entrainer plus de toxicité gastrointestinale, notamment de diarrhées avec l'irinotécan. Dans la cohorte de l'étude RETROTMZ portant sur 196 patients et décrite en 5.1, 2 patients ont arrêté le traitement pour cause de toxicité (les 2 patients étaient traités par témozolomide en association avec irinotécan); 17% des cycles ont été décalés, dont 9% pour cause de toxicité avérée.
Liste tabulée des effets indésirables
Les effets indésirables observés dans les études cliniques et rapportés depuis la commercialisation du témozolomide sont listés dans le tableau 2. Ces effets sont présentés par classe de système d'organe et par fréquence. Les groupes de fréquence sont définis selon la convention suivante : très fréquent (> 1/10) ; fréquent (> 1/100, < 1/10) ; peu fréquent (> 1/1 000, < 1/100) ; rare (> 1/10 000, < 1/1 000) ; très rare (< 1/10 000) ; fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Au sein de chaque groupe de fréquence, les effets indésirables doivent être présentés suivant un ordre décroissant de gravité.
Tableau 8. Effets indésirables chez les patients traités avec témozolomide
| Infections et infestations | |
| Fréquent : | Infections, zona, pharyngitea, candidose orale |
| Peu fréquent : | Infection opportuniste (incluant PPJ), sepsis†, méningo- encéphalite herpétique†, infection à CMV, réactivation du CMV, hépatite B†, herpès simplex, réactivation d'une infection, infection des plaies, gastro-entériteb |
| Tumeurs bénignes, malignes et non précisées | |
| Peu fréquent : | Syndrome myélodysplasique (SMD), tumeurs malignes secondaires, incluant la leucémie myéloïde |
| Affections hématologiques et du système lymphatique | |
| Fréquent : | Neutropénie fébrile, neutropénie, thrombocytopénie, lymphopénie, leucopénie, anémie |
| Peu fréquent : | Pancytopénie prolongée, anémie aplasique†, pancytopénie, pétéchie |
| Affections du système immunitaire | |
| Fréquent : | Réaction allergique |
| Peu fréquent : | Anaphylaxie |
| Affections endocriniennes | |
| Fréquent : | Syndrome cushingoïdec |
| Peu fréquent : | Diabète insipide |
| Troubles du métabolisme et de la nutrition | |
| Très fréquent : | Anorexie |
| Fréquent : | Hyperglycémie |
| Peu fréquent : | Hypokaliémie, augmentation des phosphatases alcalines |
| Affections psychiatriques | |
| Fréquent : | Agitation, amnésie, dépression, anxiété, confusion, insomnie |
| Peu fréquent : | Trouble du comportement, instabilité émotionnelle, hallucination, apathie |
| Affections du système nerveux | |
| Très fréquent : | Convulsions, hémiparésie, aphasie/dysphasie, céphalée |
| Fréquent : | Ataxie, trouble de l'équilibre, altération de la cognition, concentration altérée, baisse de conscience, étourdissements, hypoesthésie, troubles de la mémoire, troubles neurologiques, neuropathied, paresthésie, somnolence, trouble de la parole, altération du goût, tremblements |
| Peu fréquent : | Etat de mal épileptique, hémiplégie, trouble extrapyramidal, parosmie, anomalie de la démarche, hyperesthésie, trouble sensoriel, coordination anormale |
| Affections oculaires | |
| Fréquent : | Hémianopsie, vision floue, trouble de la visione, défaut du champ visuel, diplopie, douleur oculaire |
| Peu fréquent : | Acuité visuelle réduite, sécheresse oculaire |
| Affections de l'oreille et du labyrinthe | |
| Fréquent : | Surditéf, vertige, acouphène, douleur à l'oreilleg |
| Peu fréquent : | Baisse de l'audition, hyperacousie, otite moyenne |
| Affections cardiaques | |
| Peu fréquent : | Palpitation |
| Affections vasculaires | |
| Fréquent : | Hémorragie, embolie pulmonaire, thrombose veineuse profonde, hypertension |
| Peu fréquent : | Hémorragie cérébrale, bouffées vasomotrices, bouffées de chaleur |
| Affections respiratoires, thoraciques et médiastinales | |
| Fréquent : | Pneumonie, dyspnée, sinusite, bronchite, toux, infection des voies aériennes hautes |
| Peu fréquent : | Insuffisance respiratoire†, pneumopathies/pneumonie interstitielle, fibrose pulmonaire, congestion nasale |
| Affections gastro-intestinales | |
| Très fréquent : | Diarrhée, constipation, nausées, vomissements |
| Fréquent : | Stomatite, douleur abdominaleh, dyspepsie, dysphagie |
| Peu fréquent : | Distension abdominale, incontinence fécale, troubles gastro-intestinaux, hémorroïdes, bouche sèche |
| Affections hépatobiliaires | |
| Peu fréquent : | Insuffisance hépatique†, atteinte hépatique, hépatite, cholestase, hyperbilirubinémie |
| Affections de la peau et du tissu sous-cutané | |
| Très fréquent : | Rash, alopécie |
| Fréquent : | Erythème, sécheresse cutanée, prurit |
| Peu fréquent : | Nécrolyse épidermique toxique, syndrome de Stevens-Johnson, angiooedème, érythème multiforme, érythrodermie, exfoliation cutanée, réaction de photosensibilité, urticaire, exanthème, dermatite, transpiration accrue, pigmentation anormale |
| Fréquence indéterminée : | Syndrome d'hypersensibilité médicamenteuse avec éosinophilie et symptômes systémiques (DRESS) |
| Affections musculosquelettiques et systémiques | |
| Fréquent : | Myopathie, faiblesse musculaire, arthralgie, douleur dorsale, douleur musculosquelettique, myalgie |
| Affections du rein et des voies urinaires | |
| Fréquent : | Miction fréquente, incontinence urinaire |
| Peu fréquent : | Dysurie |
| Affections des organes de reproduction et du sein | |
| Peu fréquent : | Hémorragie vaginale, ménorragie, aménorrhée, vaginite, douleur mammaire, impuissance |
| Troubles généraux et anomalies au site d'administration | |
| Très fréquent : | Fatigue |
| Fréquent : | Fièvre, symptômes pseudo-grippaux, asthénie, malaise, douleur, oedème, oedème périphériquei |
| Peu fréquent : | Aggravation de l'état, raideur, oedème de la face, décoloration de la langue, soif, trouble dentaire |
| Investigations | |
| Fréquent : | Augmentation des enzymes hépatiquesj, perte de poids, prise de poids |
| Peu fréquent : | Augmentation des gamma-glutamyltransférases |
| Lésions, intoxications et complications liées aux procédures | |
| Fréquent : | Lésions radiquesk |
a Y compris pharyngite, rhinopharyngite, pharyngite à streptocoques
b Y compris gastro-entérite, gastro-entérite virale
c Y compris syndrome cushingoïde, syndrome de Cushing
d Y compris neuropathie, neuropathie périphérique, polyneuropathie, neuropathie sensitive périphérique, neuropathie motrice périphérique
e Y compris déficience visuelle, affections oculaires
f Y compris surdité, surdité bilatérale, surdité neurosensorielle, surdité unilatérale
g Y compris douleur à l'oreille, gêne au niveau de l'oreille
h Y compris douleur abdominale, douleur abdominale basse, douleur abdominale haute, gêne abdominale
i Y compris oedème périphérique, gonflement périphérique
j Y compris augmentation des tests de la fonction hépatique, augmentation de l'alanine aminotransférase, augmentation de l'aspartate aminotransférase, augmentation des enzymes hépatiques
k Y compris lésion radique, lésion radique cutanée
† Y compris des cas d'issue fatale
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé doivent déclarer tout effet indésirable suspecté selon les modalités définies dans le Protocole d'utilisation thérapeutique et de recueil de données (voir PUT RD).
SURVEILLANCE du traitement
:
1) NFS.
- avant traitement
-
3 jours précédant l'initiation du cycle suivant.
2) Fonction
hépatique :
- avant l'initiation,
- après chaque cycle.
GENOTOXICITE : Les hommes traités de ne pas procréer jusqu'à 6 mois après avoir reçu la dernière dose et de se renseigner sur la cryoconservation du sperme avant de débuter le traitement;
Les femmes en âge de procréer doivent utiliser une contraception efficace afin d'éviter toute grossesse pendant toute la durée du traitement et pendant au moins 6 mois après la fin du traitement.
Grossesse
Il n'existe aucune donnée chez la femme enceinte. Lors des études précliniques conduites chez le rat et le lapin ayant reçu une dose de 150 mg/m2 de témozolomide, des effets tératogènes et/ou toxiques pour le foetus ont été démontrés (voir rubrique Données de sécurité préclinique). Kimozo ne doit pas être administré chez la femme enceinte. Si l'administration est envisagée pendant la grossesse, la patiente doit être prévenue du risque potentiel pour le foetus.
Allaitement
On ne sait pas si le témozolomide est excrété dans le lait maternel. Par conséquent, l'allaitement doit être interrompu pendant le traitement par Kimozo.
Fertilité
Il est conseillé aux femmes en âge de procréer d'utiliser une contraception efficace afin d'éviter toute grossesse lorsqu'elles reçoivent Kimozo pendant toute la durée du traitement et pendant au moins 6 mois après la fin du traitement.
Fertilité masculine
Le témozolomide peut avoir des effets génotoxiques. Par conséquent, il est conseillé aux hommes traités par ce dernier de ne pas procréer jusqu'à 6 mois après avoir reçu la dernière dose et de se renseigner sur la cryoconservation du sperme avant de débuter le traitement, compte tenu de la possibilité d'infertilité irréversible due au témozolomide.
Les études d'interaction n'ont été réalisées que chez l'adulte.
Dans une étude de phase I, l'administration simultanée de témozolomide et de ranitidine n'a pas modifiée l'absorption du témozolomide ou l'exposition à son métabolite actif le monométhyl triazénoimidazole carboxamide (MTIC).
L'administration de témozolomide lors des repas se traduit par une diminution de 33 % de la Cmax et par une diminution de 9 % de l'aire sous la courbe (ASC). Comme on ne peut pas exclure que ce changement de la Cmax ait une signification clinique, Kimozo doit être administré en dehors des repas.
Selon une analyse pharmacocinétique de population, l'administration concomitante de dexaméthasone, de prochlorpérazine, de phénytoïne, de carbamazépine, d'ondansétron, d'antagonistes des récepteurs H2, ou de phénobarbital ne modifie pas la clairance du témozolomide. L'administration concomitante d'acide valproïque a été associée à une diminution faible mais statistiquement significative de la clairance du témozolomide.
Aucune étude n'a été réalisée pour déterminer l'effet du témozolomide sur le métabolisme ou l'élimination d'autres médicaments. Cependant, comme le témozolomide ne subit pas de métabolisme hépatique et présente une faible liaison aux protéines plasmatiques, il ne devrait pas affecter les paramètres pharmacocinétiques d'autres médicaments (voir rubrique Propriétés pharmacocinétiques).
L'utilisation de témozolomide en association avec d'autres agents myélosuppresseurs est susceptible d'accroître le risque de myélosuppression
Posologie
Kimozo doit uniquement être prescrit par des médecins qui ont l'expérience de médicaments anticancéreux en pédiatrie.
Un traitement antiémétique peut être administré (voir rubrique Mises en garde spéciales et précautions d'emploi).
Enfants et adolescents (≥ 1 an)
Traitement en monothérapie
Un cycle de traitement en monothérapie dure 28 jours. Kimozo est administré par voie orale à la dose de 150 mg/m² de surface corporelle une fois par jour pendant les 5 premiers jours du premier cycle puis le traitement est arrêté pendant les 23 jours suivants (total de 28 jours). La dose est augmentée lors du second cycle et pour les cycles suivants à 200 mg/m² de surface corporelle s'il n'y a pas de toxicité hématologique sévère (à savoir taux de polynucléaires neutrophiles (PNN) < 0,75 x 109/l et taux de plaquettes < 75 x 109/l, ou pour les patients avec atteinte de la moelle osseuse confirmée, PNN <0,50 x 109/l et plaquettes < 50 x 109/l), et si aucune toxicité non hématologique pour le cycle 1 est de Grade ≥3 selon la classification « Common Toxicity Criteria », avant l'administration du cycle suivant. Si la dose n'est pas augmentée au cycle 2, l'augmentation ne doit pas être effectuée aux cycles suivants. Une fois augmentée, la dose demeure à 200 mg/m2 par jour pour les 5 premiers jours de chaque cycle suivant à moins qu'une toxicité apparaisse. Les diminutions de dose et les arrêts de traitement doivent être effectués selon les tableaux 1 et 2.
Pendant le traitement, plusieurs bilans biologiques pourront être effectués, dont au moins une numération de la formule sanguine à réaliser dans les 3 jours précédant l'initiation du cycle suivant (entre le 25ème et le 28ème jour après la première dose de témozolomide). En fonction des résultats biologiques, le nouveau cycle peut être initié à la date prévue ou retardé, la dose peut être réduite ou encore l'administration du témozolomide interrompue selon le tableau 2
Tableau 1. Différents niveaux de dose de témozolomide pour le traitement en monothérapie
| Niveau de dose | Dose de témozolomide (mg/m2/jour) | Remarques |
| 0 | 100 | Diminution pour toxicité antérieure |
| 1 | 150 | Dose pendant le cycle 1 / Diminution pour toxicité à la dose 2 |
| 2 | 200 | Dose post cycle 1 en l'absence de toxicité |
Tableau 2. Diminution de dose de témozolomide ou arrêt pendant le traitement en monothérapie
| Toxicité | Allongement du cycle en cours | Modification de la dose à la première occurrence | Modification de la dose à la seconde occurrence |
| Toxicité hématologique | |||
| - PNN < 0,75 x 109/l a, b - Plaquettes < 75 x 109/l a b ces valeurs étant normalisées au J28 après début du cycle | Non | Pas d'adaptation de dose | Pas d'adaptation de dose |
| - PNN < 0,75 x 109/l a, b - Plaquettes < 75 x 109/l a b ces valeurs étant normalisées entre J29 et J35 après début du cycle | Oui | Pas d'adaptation de dose | Niveau de dose n-1 |
| - PNN < 0,75 x 109/l a, b - Plaquettes < 75 x 109/l a b ces valeurs étant normalisées entre J36 et J42 après début du cycle | Oui | Niveau de dose n-1 | Niveau de dose n-2 |
| - PNN < 0,75 x 109/l a, b - Plaquettes < 75 x 109/l a b valeurs non normalisées à J43 après début du cycle | Arrêt du traitement | ||
| Toxicité hépatique | |||
| Elévation des transaminases sériques (ALAT, ASAT) Grade ≥ 3 revenue à Grade ≤ 1 au J28 après début du cycle | Non | Pas d'adaptation de dose | Pas d'adaptation de dose |
| Elévation des transaminases sériques (ALAT, ASAT) Grade ≥ 3 non revenue à Grade ≤ 1 au J28 après début du cycle | Non | Niveau de dose n-1 | Arrêt du traitement |
| Autres toxicités | |||
| Autre toxicité non hématologique de Grade ≥3 non revenue à Grade ≤ 2 au J28 après début du cycle | Non | Niveau de dose n-1 | Arrêt du traitement |
a sans traitement de support (transfusion de plaquettes et/ou G-CSF) depuis au moins 3 jours
b pour les patients avec atteinte de la moelle osseuse confirmée, les seuils non acceptables de toxicité pour PNN et plaquettes sont ramenées à <0,50 x 109/l et à < 50 x 109/l respectivement.
Traitement
en association avec topotécan ou
irinotécan
Des
données sur l'utilisation du
témozolomide en association avec un
inhibiteur spécifique de
l'ADN topoisomérase I (topotécan ou
irinotécan) sont décrites à
la rubrique Propriétés
pharmacodynamiques. Les combinaisons avec topotécan ou irinotécan nécessitent
un ajustement de la dose de Kimozo et de la durée du cycle comme suit
- Un cycle de traitement en association avec topotécan dure 28 jours. Kimozo est administré par voie orale à la dose de 150 mg/m² de surface corporelle une fois par jour pendant les 5 premiers jours de chaque cycle. Topotécan est administré par voie intraveineuse sur 30 minutes à la dose de 0,75 mg/m2 de surface corporelle pendant les mêmes 5 premiers jours de chaque cycle, au moins 1 heure après l'administration du témozolomide. Puis le traitement est arrêté pendant les 23 jours suivants (total de 28 jours).
Les diminutions de dose de la polychimiothérapie et les arrêts de traitement doivent être effectués selon les tableaux 3 et 4 pour l'association témozolomide - topotécan.
- Un cycle de traitement en association avec irinotécan dure 21 jours. Kimozo est administré par voie orale à la dose de 100 mg/m² de surface corporelle une fois par jour pendant les 5 premiers jours de chaque cycle. Irinotécan est administré par voie intraveineuse sur 1 heure à la dose de 50 mg/m2 de surface corporelle pendant les mêmes 5 premiers jours de chaque cycle, au moins 1 heure après l'administration du témozolomide. Puis le traitement est arrêté pendant les 16 jours suivants (total de 21 jours).
Les diminutions de dose de la polychimiothérapie et les arrêts de traitement doivent être effectués selon les tableaux 5 et 6 pour l'association témozolomide - irinotécan.
Pendant le traitement, plusieurs bilans biologiques pourront être effectués, dont au moins une numération de la formule sanguine à réaliser dans les 3 jours précédant l'initiation du cycle suivant (entre le 18ème et le 21ème jour de traitement après la première dose de témozolomide pour l'association témozolomide - Irinotécan et entre le 25ème et le 28ème jour après la première dose de témozolomide pour l'association témozolomide - topotécan). En fonction des résultats biologiques, le nouveau cycle peut être initié à la date prévue ou retardé, les doses peuvent être réduites ou encore l'administration du traitement interrompue selon le tableau 4 pour l'association témozolomide - topotécan et le tableau 6 pour l'association témozolomide - irinotécan.
Tableau 3. Différents niveaux de dose de témozolomide et de topotécan pour le traitement en association avec topotécan
| Niveau de dose | Dose de témozolomide (mg/m2/jour) | Dose de topotécan (mg/m2/jour) | Remarques |
| 0 | 150 | 0,75 | Dose pendant le cycle 1 |
| -1 | 120 | 0,50 | Diminution pour toxicité à la dose 0 |
| -2 | 90 | 0,25 | Diminution pour toxicité à la dose -1 |
Tableau 4. Diminution de dose de témozolomide et de
topotécan ou arrêt pendant le traitement en association avec
topotécan
| Toxicité | Allongement du cycle en cours | Modification de la dose à la première occurrence | Modification de la dose à la seconde occurrence |
| Toxicité hématologique | |||
| - PNN < 0,75 x 109/l a b - Plaquettes < 75 x 109/l a b ces valeurs étant normalisées au J28 après début du cycle | Non | Pas d'adaptation de dose | Pas d'adaptation de dose |
| - PNN < 0,75 x 109/l a b - Plaquettes < 75 x 109/l a b ces valeurs étant normalisées entre J29 et J35 après début du cycle | Oui | Pas d'adaptation de dose | Témozolomide Niveau de dose -1 |
| - PNN < 0,75 x 109/l a b - Plaquettes < 75 x 109/l a b ces valeurs étant normalisées entre J36 et J42 après début du cycle | Oui | Témozolomide et topotécan Niveaux de dose -1 | Témozolomide et topotécan Niveaux de dose -2 |
| - PNN < 0,75 x 109/l a b - Plaquettes < 75 x 109/l a b valeurs non normalisées à J43 après début du cycle | Arrêt du traitement | ||
| Toxicité hépatique | |||
| Elévation des transaminases sériques (ALAT, ASAT) Grade ≥3 revenue à Grade ≤ 1 au J28 après début du cycle | Non | Pas d'adaptation de dose | Pas d'adaptation de dose |
| Elévation des transaminases sériques (ALAT, ASAT) Grade ≥3 non revenue à Grade ≤ 1 au J28 après début du cycle | Non | Témozolomide Niveau de dose -1 | Arrêt du traitement |
| Autres toxicités | |||
| Autre toxicité non hématologique de Grade ≥3 non revenue à Grade ≤ 2 au J28 après début du cycle | Non | Témozolomide et topotécan Niveaux de dose -1 | Arrêt du traitement |
a sans traitement de support (transfusion de plaquettes et/ou G-CSF) depuis au moins 3 jours
b pour les patients avec atteinte de la moelle osseuse confirmée, les seuils non acceptables de toxicité pour PNN et plaquettes sont ramenées à <0,50 x 109/l et à < 50 x 109/l respectivement.
Tableau 5. Différents niveaux de dose de témozolomide et d'irinotécan pour le traitement en association avec irinotécan
| Niveau de dose | Dose de témozolomide (mg/m2/jour) | Dose d'irinotécan (mg/m2/jour) | Remarques |
| 0 | 100 | 50 | Dose pendant le cycle 1 |
| -1 | 80 | 40 | Diminution pour toxicité à la dose 0 |
| -2 | 60 | 30 | Diminution pour toxicité à la dose - 1 |
Tableau 6. Diminution de dose de témozolomide et
d'irinotécan ou arrêt pendant le
traitement en association avec irinotécan
| Toxicité | Allongement du cycle en cours | Modification de la dose à la première occurrence | Modification de la dose à la seconde occurrence |
| Toxicité hématologique | |||
| - PNN < 0,75 x 109/l a b - Plaquettes < 75 x 109/l a b ces valeurs étant normalisées au J21 après début du cycle | Non | Pas d'adaptation de dose | Pas d'adaptation de dose |
| - PNN < 0,75 x 109/l a b - Plaquettes < 75 x 109/l a b ces valeurs étant normalisées entre J22 et J28 après début du cycle | Oui | Pas d'adaptation de dose | Témozolomide Niveau de dose -1 |
| - PNN < 0,75 x 109/l a b - Plaquettes < 75 x 109/l a b ces valeurs étant normalisées entre J29 et J35 après début du cycle | Oui | Témozolomide et irinotécan Niveaux de dose -1 | Témozolomide et irinotécan Niveaux de dose -2 |
| - PNN < 0,75 x 109/l a b - Plaquettes < 75 x 109/l a b valeurs non normalisées à J36 après début du cycle | Arrêt du traitement | ||
| Toxicité hépatique | |||
| Elévation des transaminases sériques (ALAT,ASAT) Grade ≥3 revenue à Grade ≤ 1 au J21 après début du cycle | Non | Pas d'adaptation de dose | Pas d'adaptation de dose |
| Elévation des transaminases sériques (ALAT,ASAT) Grade ≥3 non revenue à Grade ≤ 1 au J21 après début du cycle | Non | Témozolomide Niveau de dose -1 | Arrêt du traitement |
| Autres toxicités | |||
| Diarrhée de Grade 3 et 4 >3 jours malgré un traitement adapté avec du lopéramide | Non | Irinotécan Niveau de dose -1 Si le même niveau de toxicité persiste > 2 semaines malgré traitement symptomatique adapté, arrêt du traitement | Irinotécan Niveau de dose -2 Si le même niveau de toxicité persiste > 2 semaines malgré traitement symptomatique adapté, arrêt du traitement |
| Oui | Irinotécan Niveau de dose -1 Si diarrhée en cours au Jour 21, retarder le cycle suivant jusqu'à 2 semaines tant que la diarrhée n'est pas revenue à Grade ≤ 1 Si diarrhée ne s'améliore pas en 2 semaines, arrêt du traitement | Irinotécan Niveau de dose -2 Si diarrhée en cours au Jour 21, retarder le cycle suivant jusqu'à 2 semaines tant que la diarrhée n'est pas revenue à Grade ≤ 1 Si diarrhée ne s'améliore pas en 2 semaines, arrêt du traitement | |
| Autre toxicité non hématologique de Grade ≥3 non revenue à Grade ≤ 2 au J28 après début du cycle | Non | Témozolomide et irinotécan Niveau de dose -1 | Arrêt du traitement |
a sans traitement de support (transfusion de plaquettes et/ou G-CSF) depuis au moins 3 jours
b pour les patients avec atteinte de la moelle osseuse confirmée, les seuils non acceptables de toxicité pour PNN et plaquettes sont ramenées à <0,50 x 109/l et à < 50 x 109/l respectivement
Patients atteints d'une insuffisance hépatique ou rénale
Les paramètres pharmacocinétiques du témozolomide sont comparables chez les patients ayant une fonction hépatique normale et chez ceux qui sont atteints d'une insuffisance hépatique faible ou modérée. Aucune donnée n'est disponible concernant l'administration de témozolomide chez les patients atteints d'insuffisance hépatique sévère (stade C de la classification de Child) ou d'insuffisance rénale. Sur la base des propriétés pharmacocinétiques du témozolomide, il est peu probable qu'une réduction de dose soit nécessaire chez les patients atteints d'insuffisance hépatique sévère ou d'insuffisance rénale quelqu'en soit le degré. Cependant, des précautions doivent être prises lorsque le témozolomide est administré chez ces patients.
Durée du traitement
Le traitement par Kimozo peut être poursuivi jusqu'à progression objective de la maladie ou apparition d'une toxicité inacceptable.
Mode d'administration
Kimozo est administré par voie orale et doit être agité vigoureusement afin d'être parfaitement réhomogénéisé avant administration.
Kimozo doit être pris à jeun, en utilisant la seringue de dosage graduée de 5 mL fournie. La seringue mise à disposition est marquée de 0,5 ml à 5 mL et a une précision de 0,1 mL.
Pour les doses journalières totales supérieures à 200 mg (volume de suspension supérieur à 5 mL), la seringue doit être remplie deux fois, le volume de remplissage a minima de la seringue étant de 0,5 mL.
Pour une dose à administrer de 5,4 mL (correspondant à une dose journalière totale de 216 mg de témozolomide nécessaire pour traiter un patient ayant une surface corporelle de 1,06 m2 à la dose de 200 mg/m2 selon le tableau 7), il est recommandé d'administrer 2 volumes équivalents de 2,7 mL.
Il est recommandé au professionnel de santé de conseiller le patient
ou la personne soignante à utiliser la seringue de dosage pour administrer le
volume correct, correspondant à la dose prescrite.
Le pharmacien dispensant le médicament veillera à préciser la dose
journalière totale en volume (mL) à
administrer au patient et
le nombre de prises (une prise si
≤ 5 mL ou
deux si > 5
mL) et le
volume en mL de chaque
prise, sur la boite de Kimozo à l'endroit prévu à cet effet.
Tableau 7. Correspondance entre la surface corporelle (SC), la dose journalière et le volume de Kimozo suspension buvable à administrer aux patients pédiatriques
| 100mg/m² | 150mg/m² | 200mg/m² | ||||
| SC (m²) | Dose (mg) | Volume (ml) | Dose (mg) | Volume (ml) | Dose (mg) | Volume (ml) |
| 0,27-0,30 | 28 | 0,7 | 44 | 1,1 | 56 | 1,4 |
| 0,31-0,34 | 32 | 0,8 | 48 | 1,2 | 64 | 1,6 |
| 0,35-0,38 | 36 | 0,9 | 56 | 1,4 | 72 | 1,8 |
| 0,39-0,42 | 40 | 1,0 | 60 | 1,5 | 80 | 2,0 |
| 0,43-0,46 | 44 | 1,1 | 68 | 1,7 | 88 | 2,2 |
| 0,47-0,50 | 48 | 1,2 | 72 | 1,8 | 96 | 2,4 |
| 0,51-0,54 | 52 | 1,3 | 80 | 2,0 | 104 | 2,6 |
| 0,55-0,58 | 56 | 1,4 | 84 | 2,1 | 112 | 2,8 |
| 0,59-0,62 | 60 | 1,5 | 92 | 2,3 | 120 | 3,0 |
| 0,63-0,66 | 64 | 1,6 | 96 | 2,4 | 128 | 3,2 |
| 0,67-0,70 | 68 | 1,7 | 104 | 2,6 | 136 | 3,4 |
| 0,71-0,74 | 72 | 1,8 | 108 | 2,7 | 144 | 3,6 |
| 0,75-0,78 | 76 | 1,9 | 116 | 2,9 | 152 | 3,8 |
| 0,79-0,82 | 80 | 2,0 | 120 | 3,0 | 160 | 4,0 |
| 0,83-0,86 | 84 | 2,1 | 128 | 3,2 | 168 | 4,2 |
| 0,87-0,90 | 88 | 2,2 | 132 | 3,3 | 176 | 4,4 |
| 0,91-0,94 | 92 | 2,3 | 140 | 3,5 | 184 | 4,6 |
| 0,95-0,98 | 96 | 2,4 | 144 | 3,6 | 192 | 4,8 |
| 0,99-1,02 | 100 | 2,5 | 152 | 3,8 | 200 | 5,0 |
| 1,03-1,06 | 104 | 2,6 | 156 | 3,9 | 208 | 5,2 |
| 1,07-1,10 | 108 | 2,7 | 164 | 4,1 | 216 | 5,4 |
| 1,11-1,14 | 112 | 2,8 | 168 | 4,2 | 224 | 5,6 |
| 1,15-1,18 | 116 | 2,9 | 176 | 4,4 | 232 | 5,8 |
| 1,19-1,22 | 120 | 3,0 | 180 | 4,5 | 240 | 6,0 |
| 1,23-1,26 | 124 | 3,1 | 188 | 4,7 | 248 | 6,2 |
| 1,27-1,30 | 128 | 3,2 | 192 | 4,8 | 256 | 6,4 |
| 1,31-1,34 | 132 | 3,3 | 200 | 5,0 | 264 | 6,6 |
| 1,35-1,38 | 136 | 3,4 | 204 | 5,1 | 272 | 6,8 |
| 1,39-1,42 | 140 | 3,5 | 212 | 5,3 | 280 | 7,0 |
| 1,43-1,46 | 144 | 3,6 | 216 | 5,4 | 288 | 7,2 |
| 1,47-1,50 | 148 | 3,7 | 224 | 5,6 | 296 | 7,4 |
| 1,51-1,54 | 152 | 3,8 | 228 | 5,7 | 304 | 7,6 |
| 1,55-1,58 | 156 | 3,9 | 236 | 5,9 | 312 | 7,8 |
| 1,59-1,62 | 160 | 4,0 | 240 | 6,0 | 320 | 8,0 |
| 1,63-1,66 | 164 | 4,1 | 248 | 6,2 | 328 | 8,2 |
| 1,67-1,70 | 168 | 4,2 | 252 | 6,3 | 336 | 8,4 |
| 1,71-1,74 | 172 | 4,3 | 260 | 6,5 | 344 | 8,6 |
| 1,75-1,78 | 176 | 4,4 | 264 | 6,6 | 352 | 8,8 |
| 1,79-1,82 | 180 | 4,5 | 272 | 6,8 | 360 | 9,0 |
| 1,83-1,86 | 184 | 4,6 | 276 | 6,9 | 368 | 9,2 |
| 1,87-1,90 | 188 | 4,7 | 284 | 7,1 | 376 | 9,4 |
| 1,91-1,94 | 192 | 4,8 | 288 | 7,2 | 384 | 9,6 |
| 1,95-1,98 | 196 | 4,9 | 296 | 7,4 | 392 | 9,8 |
| 1,99-2,02 | 200 | 5,0 | 300 | 7,5 | 400 | 10,0 |
Après l'administration de Kimozo, il convient de boire un verre ou
quelques gorgées d'eau ou de jus de
fruit afin de se rincer la bouche (voir rubrique Mises en garde spéciales et
précautions d'emploi).
Si des vomissements surviennent après l'administration de la dose, ne pas administrer une deuxième dose le même jour et attendre la prochaine dose prévue.
Durée de conservation :
Avant ouverture : 2 ans.
Après ouverture : le produit doit être utilisé dans les 5 jours.
Précautions particulières de conservation :
A conserver au réfrigérateur (entre 2°C et 8°C)
Conserver le flacon soigneusement fermé (voir rubrique Précautions particulières d'élimination et de manipulation).
Sans objet.
Les doses de 500, 750, 1 000 et 1 250 mg/m2 (dose totale par cycle de 5 jours) ont été évaluées cliniquement chez les patients adultes. La toxicité dose-limitante était la toxicité hématologique et était rapportée avec toute dose mais attendue pour être plus sévère aux doses supérieures. Un surdosage de 10 000 mg (dose totale pour un seul cycle, sur 5 jours) a été constaté chez un patient et les effets indésirables rapportés étaient pancytopénie, pyrexie, défaillance de multiples organes et décès. Chez des patients ayant pris la dose recommandée pendant plus de 5 jours de traitement (jusqu'à 64 jours) les effets indésirables incluaient une aplasie médullaire, avec ou sans infection, dans certains cas sévère et prolongée et pouvant entraîner un décès. En cas de surdosage, un bilan hématologique est nécessaire. Des soins intensifs doivent être mis en place si nécessaire.
Classe pharmacothérapeutique : Agents antinéoplasiques - Autres agents alkylants, code ATC : L01AX03
Mécanisme d'action
Le témozolomide est un dérivé triazène qui subit une conversion chimique rapide à pH physiologique en monométhyl triazenoimidazole carboxamide (MTIC) actif. La cytotoxicité du MTIC est vraisemblablement due principalement à une alkylation de l'ADN au niveau de la guanine en position O6 et à une alkylation supplémentaire en position N7. Les lésions cytotoxiques qui sont développées par la suite sont supposées entraîner une réparation aberrante de l'ADN méthylé
Efficacité et sécurité clinique dans la population pédiatrique
Neuroblastome réfractaire ou en rechute (monothérapie)
Etude de phase II dans le neuroblastome réfractaire ou en rechute (Rubie 2006)
25 patients avec un neuroblastome de haut risque (maladie métastatique ou localisée avec amplification de NMYC), parmi lesquels 10 patients avec maladie réfractaire et 15 patients avec récidive, ont été traités par du témozolomide en monothérapie. Les patients étaient âgés de 1,7 à 15,9 ans, avec un âge médian de 6,5 ans. La posologie du témozolomide était de 150 à 200 mg/m2/jour les jours 1 - 5 de chaque cycle de 28 jours jusqu'à la progression ou un maximum de 12 cycles. 20% des patients (n=5) ont présenté une réponse au témozolomide à l'imagerie (1 très bonne réponse et 4 réponses partielles) et 68% (n=17) ont présenté une absence de progression (réponse complète ou partielle, réponse mixte, stabilité de la maladie).
Etude compassionnelle dans les tumeurs solides pédiatriques résistantes ou réfractaires (De Sio 2006)
52 patients ont été traités par du témozolomide en monothérapie. 17 patients étaient atteints d'un neuroblastome de haut risque réfractaire ou en rechute et âgés de 2 à 16,9 ans avec un âge médian de 7,8 ans. La posologie du témozolomide était de 180 à 215 mg/m²/jour, les jours 1 - 5 de chaque cycle de 28 jours jusqu'à la progression ou un maximum de 24 cycles. Parmi les 17 patients atteints d'un neuroblastome, 12% des patients (n=2) ont présenté une réponse au témozolomide à l'imagerie (1 réponse complète et 1 réponse mineure) et 65% (n=11) ont présenté une absence de progression (réponse complète ou partielle ou mineure, réponse mixte, stabilité de la maladie).
Etude de phase II dans le neuroblastome réfractaire ou en rechute (Etude BEACON en cours dont lesdonnées fournies ci-dessous sont préliminaires, Moreno 2019)
61 patients âgés de 1 à 18 ans (âge médian de 5,9 ans) et atteints d'un neuroblastome de haut risque réfractaire ou en rechute ont été traités avec du témozolomide en monothérapie ou en association avec bevacizumab. La posologie du témozolomide était de 200 mg/m²/jour, les jours 1 - 5 de chaque cycle de 28 jours jusqu'à 6 cycles ou un maximum de 12 cycles si bénéfice clinique. 59 patients ont été traités pendant 3,7 cycles en moyenne. 24% des patients (n=14) ont présenté une réponse au témozolomide (1 réponse complète et 13 réponses partielles) et 63% (n=37) ont présenté une absence de progression (réponse complète ou partielle ou mineure, stabilité de la maladie).
Neuroblastome réfractaire ou en rechute (association avec irinotécan)
Etude de phase I dans les tumeurs solides pédiatriques résistantes ou réfractaires (Wagner 2004)
12 patients âgés de 1 à 23 ans avec un âge médian de 12,5 ans ont été traités par du témozolomide combiné à de l'irinotecan. 2 patients étaient atteints d'un neuroblastome de haut risque réfractaire ou en rechute. La posologie du témozolomide était de 100 mg/m2/jour, les jours 1 - 5 de chaque cycle de 28 jours, celle de l'irinotecan était de 10 ou 15mg/m2/jour, les jours 1-5 et 8-12 de chaque cycle de 28 jours, jusqu'à progression ou un maximum de 17 cycles. Parmi les 2 patients atteints d'un neuroblastome, 1 patient a présenté une réponse partielle et 1 patient a présenté une maladie stabilisée.
Etude dans le neuroblastome réfractaire ou en rechute (Kushner 2006)
49 patients âgés de 2,3 à 25,9 ans (âge médian : 5,5 ans) et présentant un neuroblastome de haut risque réfractaire ou en rechute ont été traités par du témozolomide combiné à de l'irinotécan. La posologie du témozolomide était de 150 mg/m2/jour les jours 1 - 5 de chaque cycle de 21 ou 28 jours, celle de l'irinotecan était de 50mg/m2/jour les jours 1-5 de chaque cycle de 21 ou 28 jours, jusqu'à progression. Parmi les 36 patients évaluables pour l'efficacité, 8% ont répondu (2 patients ont présenté une réponse complète et 1 patient une réponse partielle) et 75% (n=27) ont présenté une absence de progression (9 patients une réponse objective mineure et 15 patients une maladie stabilisée).
Etude de phase I dans le neuroblastome réfractaire ou en rechute (Wagner 2009)
14 patients âgés de 3 à 22 ans (âge médian : 7 ans) et présentant un neuroblastome de haut risque réfractaire ou en rechute ont été traités par du témozolomide combiné à de l'irinotécan. La posologie du témozolomide était de 75 ou 100 mg/m2/jour les jours 1 - 5 de chaque cycle de 21 jours, celle de l'irinotecan était de 30 ou 60 mg/m2/jour les jours 1-5 et 8-12 de chaque cycle de 21 jours, jusqu'à progression. 1 patient (7%) a répondu et 50% (n=7) ont présenté une absence de progression (1 réponse complète et 6 patients avec maladie stabilisée).
Etude de phase II dans le neuroblastome réfractaire ou en rechute (Bagatell 2011)
55 patients présentant un neuroblastome réfractaire ou en rechute ont été traités par du témozolomide combiné à de l'irinotécan. Les patients étaient âgés de 3 à 22 ans, avec un âge médian de 7 ans. La posologie du témozolomide était de 100 mg/m2/jour les jours 1 - 5 de chaque cycle de 21 jours, celle de l'irinotecan était de 10 mg/m2/jour les jours 1-5 et 8-12 de chaque cycle de 21 jours, jusqu'à progression avec un maximum de 6 cycles. Sur les 54 patients évalués pour l'efficacité, 15% des patients (n=8) ont répondu au traitement (4 réponses complètes et 4 réponses partielles) et 69% (n=37) ont présenté une absence de progression (4 réponses complètes, 4 réponses partielles et 29 maladies stabilisées).
Etude de phase II dans le neuroblastome réfractaire ou en rechute (Etude BEACON en cours dont lesdonnées fournies ci-dessous sont préliminaires, Moreno 2019)
60 patients âgés de 1 à 22 ans avec un âge médian de 5,3 ans et atteints d'un neuroblastome de haut risque réfractaire ou en rechute ont été inclus pour traitement avec témozolomide combiné à de l'irinotécan, ou combiné à de l'irinotécan et du bevacizumab. La posologie du témozolomide était de 150 mg/m2/jour, les jours 1 - 5 de chaque cycle de 21 jours, celle de l'irinotecan était de 50 mg/m2/jour les jours 1-5 de chaque cycle de 21 jours jusqu'à 6 cycles ou un maximum de 12 cycles si bénéfice clinique. 58 patients ont été traités pendant 4,5 cycles en moyenne. 17% des patients (n=10) ont présenté une réponse au témozolomide (3 réponses complètes et 7 réponses partielles) et 77% (n=46) ont présenté une absence de progression (réponse complète ou partielle ou mineure, stabilité de la maladie).
Neuroblastome réfractaire ou en rechute (association avec topotécan)
Etude de phase I dans les tumeurs solides pédiatriques résistantes ou réfractaires (Rubie 2010)
16 patients âgés de 3 à 19 ans avec un âge médian de 8,5 ans ont été traités par du témozolomide combiné à du topotécan. 8 patients étaient atteints d'un neuroblastome réfractaire ou en rechute. La posologie du témozolomide était de 100 ou 150 mg/m2/jour, les jours 1 - 5 de chaque cycle de 28 jours, celle du topotécan était de 0,75 ou 1 mg/m2/jour les jours 1-5 de chaque cycle de 28 jours, jusqu'à progression ou un maximum de 12 cycles. Parmi les 8 patients atteints d'un neuroblastome, 3 patients (38%) ont répondu au traitement, dont 1 patient avec une réponse complète, 1 patient avec une réponse partielle puis complète après chirurgie et 1 patient une réponse partielle. Tous les patients (100%, n=8) ont présenté une absence de progression (2 réponses complètes, 1 réponse partielle et 5 maladies stabilisées).
Etude de phase II dans le neuroblastome réfractaire ou en rechute (Di Giannatale 2014)
38 patients présentant un neuroblastome de haut risque réfractaire ou en rechute ont été traités par du témozolomide combiné à du topotécan. Les patients étaient âgés de 1 à 19,8 ans, avec un âge médian de 5,4 ans. La posologie du témozolomide était de 150 mg/m2/jour les jours 1 - 5 de chaque cycle de 28 jours, celle du topotécan était de 0,75 mg/m2/jour les jours 1-5 de chaque cycle de 28 jours, jusqu'à progression ou un maximum de 12 cycles. Sur les 38 patients évalués pour l'efficacité, 24% des patients (n=9) ont répondu au traitement (3 réponses complètes et 6 réponses partielles) et 79% (n=30) ont présenté une absence de progression (3 réponses complètes, 6 réponses partielles, 4 réponses mixtes et 17 maladies stabilisées).
Données rétrospectives d'utilisation en vie réelle dans le traitement du neuroblastomeréfractaire ou en rechute (étude RETROTMZ)
Cette étude rétrospective rapporte les données de 196 patients pédiatriques présentant un neuroblastome réfractaire ou en rechute et traités à partir de 2004 jusqu'en 2021 par du témozolomide en monothérapie ou en association. Les patients étaient âgés de 1 à 18 ans, avec un âge médian de 4 ans. La majorité des patients ont été traités par du témozolomide associé au topotécan (41%, n=81), par du témozolomide seul (30%, n=59) ou associé à l'irinotécan (20%, n=39).
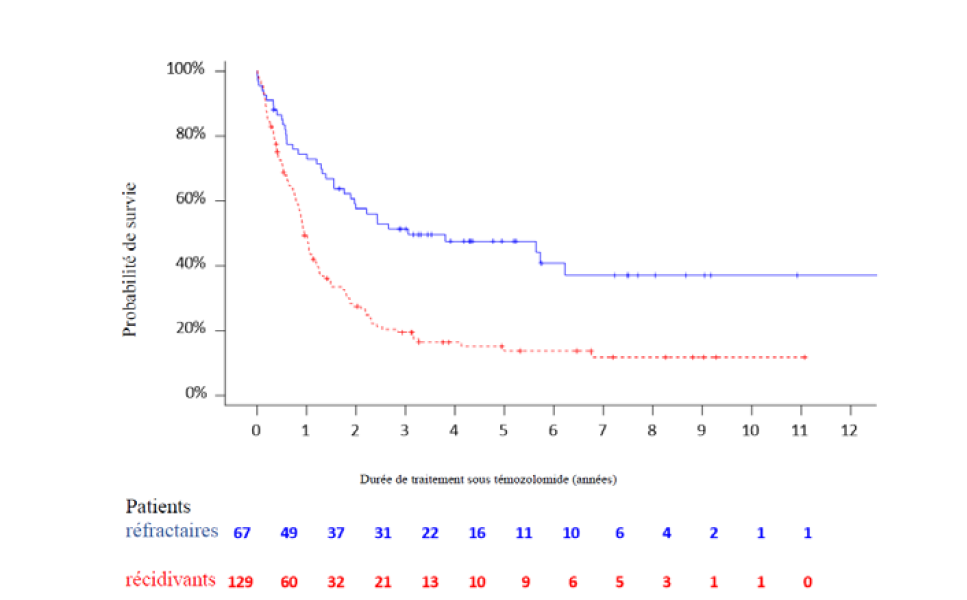
Les résultats globaux d'efficacité sont résumés dans les tableaux 9 et 10 et la figure 1.
Les résultats d'efficacité selon le schéma thérapeutique (témozolomide en monothérapie ou en association avec irinotécan et topotécan) sont résumés dans les tableaux 11, 12 et 13 respectivement.
Tableau 9 : Taux de réponse global dans l'étude RETROTMZ (témozolomide monothérapie et associations)
| | |||
| | Cure initiale de témozolomide | Cures complémentaires de témozolomide | |
| | Patients réfractaires | Patients récidivants | Patients récidivants |
| Patients avec évaluation formelle (n) | 56 | 95 | 23 |
| Nombre de répondeurs, n (%) | 28 (50) | 45 (47) | 10 (43) |
| - Réponse complète, n (%) | 3 (5) | 13 (14) | 3 (13) |
| - Réponse complète maintenue1, n (%) | | 2 (2) | 6 (26) |
| - Réponse partielle, n (%) | 25 (45) | 29 (31) | 1 (4) |
| - Réponse partielle maintenue1, n (%) | | 1 (1) | |
| Maladie stabilisée, n (%) | 19 (34) | 27 (28) | 3 (13) |
1 réponse obtenue par un traitement différent et maintenue sous traitement avec témozolomide
Tableau 10 : Survie globale (OS) à 1, 2 et 5 ans dans l'étude RETROTMZ (témozolomide monothérapie et associations)
| Survie globale [95% IC] | Patients réfractaires (n= 67) | Patients récidivants (n=129) |
| 1 an | 74.4% [63.9 ; 84.9] | 49.3% [40.6 ; 58.1] |
| 2 ans | 57.5% [45.6 ; 69.5] | 27.5% [19.5 ; 35.4] |
| 5 ans | 47.5% [35.2 ; 59.9] | 13.8% [7.1 ; 20.4] |
Figure 1 : Survie globale dans l'étude RETROTMZ (témozolomide monothérapie et associations)
Témozolomide monothérapie
Tableau 11 : Taux de réponse global dans l'étude RETROTMZ (témozolomide monothérapie, cure initiale)
| | Patients réfractaires | Patients récidivants |
| Patients avec évaluation formelle (n) | 13 | 21 |
| Nombre de répondeurs, n (%) | 5 (38,5) | 6 (28,6) |
| - Réponse complète, n (%) | 0 | 1 (4,8) |
| - Réponse complète maintenue1, n (%) | 0 | 0 |
| - Réponse partielle, n (%) | 5 (38,5) | 4 (19,0) |
| - Réponse partielle maintenue1, n (%) | 0 | 1 (4,8) |
| Maladie stabilisée, n (%) | 3 (23,1) | 8 (38,1) |
1 réponse obtenue par un traitement différent et maintenue sous traitement avec témozolomide
Survie globale (OS) médiane dans l'étude RETROTMZ (témozolomide monothérapie)
Patients réfractaires [95% CI]: 1,21 ans [0,33;2,43]
Patients récidivants [95% CI]: 0,73 ans [0,42;1,01]
Témozolomide et irinotécan
Tableau 12 : Taux de réponse global dans l'étude RETROTMZ (témozolomide et irinotécan, cure initiale)
| | Patients réfractaires | Patients récidivants |
| Patients avec évaluation formelle (n) | 10 | 20 |
| Nombre de répondeurs, n (%) | 5 (50) | 10 (50) |
| - Réponse complète, n (%) | 1 (10) | 3 (15) |
| - Réponse complète maintenue1, n (%) | 0 | 1 (5) |
| - Réponse partielle, n (%) | 4 (40) | 6 (30) |
| - Réponse partielle maintenue1, n (%) | 0 | 0 |
| Maladie stabilisée, n (%) | 2 (20) | 6 (30) |
1 réponse obtenue par un traitement différent et maintenue sous traitement avec témozolomide
Survie globale (OS) médiane dans l'étude RETROTMZ (témozolomide et irinotécan)
Patients réfractaires [95% CI]: 1,55 ans [0,1;ND*] Patients récidivants [95% CI]: 0,91 ans [0,76;2,2] ND*: non déterminable
Témozolomide et topotécan
Tableau 13 : Taux de réponse global dans l'étude RETROTMZ (témozolomide et topotécan, cure initiale)
| | Patients réfractaires | Patients récidivants |
| Patients avec évaluation formelle (n) | 27 | 44 |
| Nombre de répondeurs, n (%) | 16 (59,3) | 26 (59,1) |
| - Réponse complète, n (%) | 2 (7,4) | 7 (15,9) |
| - Réponse complète maintenue1, n (%) | 0 | 1 (2,3) |
| - Réponse partielle, n (%) | 14 (51,9) | 18 (40,9) |
| - Réponse partielle maintenue1, n (%) | 0 | 0 |
| Maladie stabilisée, n (%) | 10 (37,0) | 9 (20,5) |
1 réponse obtenue par un traitement différent et maintenue sous traitement avec témozolomide
Survie globale (OS) médiane dans l'étude RETROTMZ (témozolomide et topotécan)
Patients réfractaires [95% CI]: ND* [3,05;ND*] Patients récidivants [95% CI]: 1,25 ans [0,79;1,87] ND*: non déterminable
Le témozolomide est spontanément hydrolysé au pH physiologique, principalement en son métabolite actif, le 3- méthyl-(triazen-1-yl)-imidazole-4-carboxamide (MTIC). Le MTIC est spontanément hydrolysé en 5-amino-imidazole-4-carboxamide (AIC), un intermédiaire connu dans la biosynthèse de la purine et de l'acide nucléique, et en diazométhane, métabolite actif alkylant supposé. Il semble que la cytotoxicité du MTIC soit principalement due à l'alkylation de l'ADN essentiellement aux positions O6 et N7 de la guanine. Concernant l'Aire Sous la Courbe (ASC) du témozolomide, l'exposition aux MTIC et AIC est de ~ 2,4 % et 23 %, respectivement. In vivo, le t1/2 du MTIC était similaire à celui du témozolomide, 1,8 heures.
Absorption
Après administration orale chez l'adulte, le témozolomide est rapidement absorbé avec des pics de concentration plasmatique observés parfois dès 20 minutes après administration (temps moyen compris entre 0,5 et 1,5 heures). Après administration orale de témozolomide marqué au 14C, l'excrétion fécale moyenne de 14C dans les 7 jours suivant l'administration était de 0,8 %, signe d'une absorption complète.
Distribution
Le témozolomide est faiblement lié aux protéines plasmatiques (10 à 20 %), et par conséquent, est peu susceptible d'interagir avec des produits très fortement liés aux protéines.
Les études cliniques utilisant la technique de TEP (Tomographie par Emission de Positrons) et les données précliniques suggèrent que le témozolomide traverse rapidement la barrière hémato- encéphalique et pénètre dans le liquide céphalo-rachidien (LCR). La pénétration dans le LCR a été confirmée chez un patient ; sur la base de l'ASC du témozolomide, l'exposition du LCR était approximativement 30 % de celle du plasma, ce qui est cohérent avec les données chez l'animal.
Élimination
La demi-vie plasmatique (t½) est approximativement de 1,8 heures. La voie principale d'élimination du C14 est la voie rénale. Après administration orale, environ 5 à 10 % de la dose sont retrouvés sous forme inchangée dans les urines dans les 24 heures, et le reste est excrété sous forme d'acide témozolomide, de 5-aminoimidazole-4-carboxamide (AIC) ou de métabolites polaires non identifiés.
L'augmentation des concentrations plasmatiques est dose-dépendante. La clairance plasmatique, le volume de distribution et la demi-vie sont indépendants de la dose.
Populations spéciales
L'analyse des paramètres pharmacocinétiques de population du témozolomide a montré que la clairance plasmatique du témozolomide est indépendante de l'âge, de la fonction rénale ou de la consommation de tabac. Dans une autre étude pharmacocinétique, les profils pharmacocinétiques plasmatiques des patients atteints d'insuffisance hépatique faible à modéré étaient identiques à ceux observés chez les patients dont la fonction hépatique était normale.
Les enfants présentent une ASC plus élevée que les adultes ; cependant, la dose maximale tolérée (DMT) est de 1 000 mg/m2 par cycle de traitement à la fois chez l'enfant et chez l'adulte
Le témozolomide a une influence mineure sur l'aptitude à réaliser des activités dangereuses en raison de la fatigue et de la somnolence (voir rubrique Effets indésirables). Il convient de surveiller attentivement les jeunes patients qui font du vélo, de l'escalade ou toute autre activité dangereuse.
Des études de toxicité à cycle unique (5 jours de traitement, 23 jours sans traitement), à 3 et 6 cycles ont été réalisées chez le rat et le chien. Les principales cibles de la toxicité ont été la moelle osseuse, le système lymphoréticulaire, les testicules, le tractus gastro-intestinal et, pour des doses plus fortes, létales chez 60 % à 100 % des rats et des chiens testés, une dégénérescence rétinienne est survenue. La plupart des phénomènes toxiques ont été réversibles, à l'exception des effets secondaires sur le système de reproduction mâle et de la dégénérescence rétinienne. Cependant, comme les doses impliquées dans cette dégénérescence appartiennent à l'intervalle de doses létales, et qu'aucun effet semblable n'a été observé lors des études cliniques, ce résultat n'a pas de signification clinique.
Le témozolomide est un agent alkylant embryotoxique, tératogène et génotoxique. Le témozolomide est plus toxique chez le rat et le chien que chez l'homme, et la dose thérapeutique est proche de la dose létale minimale chez le rat et le chien. Les diminutions dose-dépendantes du nombre des leucocytes et des plaquettes apparaissent comme des indicateurs sensibles de la toxicité. Différents types de néoplasmes ont été observés lors de l'étude de toxicité après 6 cycles de traitement chez le rat, dont carcinome mammaire, kérato-acanthome cutané et adénome baso-cellulaire alors qu'aucune tumeur, ni aucun changement prénéoplasique n'ont été observés au cours des études chez le chien. Le rat semble être particulièrement sensible aux effets oncogènes du témozolomide, avec l'apparition des premières tumeurs dans les 3 mois suivant le début du traitement. La période de latence est très courte, même pour un agent alkylant.
Les résultats des tests d'Ames/salmonella et d'aberration chromosomique sur lymphocyte humain démontrent l'existence d'un potentiel mutagène.
Toute personne manipulant Kimozo doit se laver les mains avant et après l'administration d'une dose. Pour réduire le risque d'exposition, il est recommandé que les parents et les personnes soignantes portent des gants à usage unique pour manipuler Kimozo.
Tout contact de Kimozo avec la peau ou une membrane muqueuse doit être évité. Si Kimozo entre en contact avec la peau ou une muqueuse, laver la zone concernée immédiatement et abondamment à grande eau et au savon. Tout produit renversé doit être essuyé immédiatement.
Avant chaque utilisation, la suspension de Kimozo doit être homogénéisée en agitant le flacon vigoureusement pendant 10 secondes puis par retournement pendant 10 secondes. Cette opération est répétée trois fois. Après inspection visuelle et contrôle de l'homogénéité de la suspension, la seringue orale est insérée dans l'adaptateur du bouchon prévu à cet effet.
Le volume de suspension nécessaire au traitement est prélevé à l'aide de la seringue de dosage graduée fournie, en une ou deux fois conformément à la posologie prescrite (voir section Posologie et mode d'administration tableau de correspondance entre la surface corporelle du patient, la dose journalière de témozolomide et le volume de suspension à administrer).
Utilisation dans la population pédiatrique
Kimozo est administré pur sans dilution préalable. Kimozo est administré oralement au patient en position assise et en positionnant l'extrémité de la seringue dans le creux de la joue. Après chaque administration de Kimozo, il convient de boire quelques gorgées ou un petit verre d'eau ou de jus de fruit, afin de rincer la bouche de toute trace de Kimozo.
Les flacons de Kimozo seront tenus strictement hors de la vue et de la portée des enfants. L'ingestion accidentelle peut être fatale pour les enfants.
Conserver le flacon soigneusement fermé pour protéger l'intégrité du produit et minimiser le risque de déversement accidentel.
Elimination
Kimozo est un cytotoxique.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I
Médicament soumis à prescription hospitalière.
Prescription réservée aux spécialistes en oncologie ou aux médecins compétents en cancérologie.
Médicament nécessitant une surveillance particulière pendant le traitement.
Suspension buvable.
Suspension homogène blanche à légèrement rosée ou beige.
20 ml de suspension buvable, homogène blanche à légèrement rosée ou beige, dans un flacon en polyéthylène-téréphtalate (PET) de 30 ml avec un bouchon sécurité-enfant et un insert pour seringue de dosage pour administration orale.
Chaque boite de Kimozo contient 1 flacon de suspension buvable et 1 seringue de dosage graduée jusqu'à 5 ml.
Chaque flacon de Kimozo contient 20 ml de suspension buvable à 40 mg/ml, soit 800 mg de témozolomide.
Excipient à effet notoire
Ce médicament contient 1 mg/ml de benzoate de sodium (E211). Pour la liste complète des excipients, voir rubrique Liste des excipients.
Gomme xanthane
Acide citrique
Dioxyde de silicium amorphe
Sucralose
Arôme Cola
Benzoate de sodium (E211)
Eau purifiée